研究论文/Research Article
利用双向电泳方法初步分析大豆根、茎、叶蛋白质特点 
2. 昆明学院生命科学与技术系, 昆明, 650214
3. 云南省农业科学院生物技术与种质资源研究所, 昆明, 650223
*同等贡献作者
 作者
作者  通讯作者
通讯作者
《分子植物育种》网络版, 2014 年, 第 12 卷, 第 2 篇 doi: 10.5376/mpb.cn.2014.12.0002
收稿日期: 2013年12月25日 接受日期: 2014年01月05日 发表日期: 2014年02月27日
引用格式(中文):
董玉梅等, 2014, 利用双向电泳方法初步分析大豆根、茎、叶蛋白质特点, 分子植物育种(online), 12(2): 1008-1015 (doi: 10.5376/mpb.cn.2014.12.0002)
引用格式(英文):
Dong et al., 2014, Characters of Root, Stem and Leaf Proteins in Soybean Analyzed Using a 2-DE Approach, Fenzi Zhiwu Yuzhong (online) (Molecular Plant Breeding), 12(2): 1008-1015 (doi: 10.5376/mpb.cn.2014.12.0002)
本研究采TCA丙酮沉淀法提取大豆根、茎、叶蛋白质,用预制胶条(IPG, pH 4~7)进行双向电泳。根据电泳结果,初步分析了大豆根、茎、叶蛋白质的等电点和分子量组成特点如下:等电点在4~5间所占百分率最高的是大豆茎蛋白(35%),等电点在5~6间所占百分率最高的是大豆叶蛋白(52%),等电点在6~7间所占百分率最高的是大豆根蛋白(34%);而小于18.4 kD的小分子量蛋白以及大于75 kD的大分子量蛋白都是根器官中蛋白所占百分率最高,不同器官蛋白质的组成不同,反映了这些器官在结构和功能上存在差异的分子基础。
通常,蛋白质组学(Proteomics)的研究策略有两种:一是着重研究不同生长状态或病理情况下蛋白质的差异表达情况,进而揭示细胞对生物胁迫或非生物胁迫的生理和病理状态进程、调控机制等,同时获得对某些关键蛋白的定性和定量分析;二是采用高通量的蛋白测序及相关技术阐明某特定生物体、组织、甚至是细胞器内尽可能多的蛋白质以了解其发育特征(Hakeem et al., 2012; Colmsee et al., 2012; Que et al., 2011; 刘艳艳, 2009)。因此,蛋白质组学技术是目前解析植物应答各种胁迫(包括非生物或病菌侵染等在内的各种胁迫)的分子机制的有力工具(Yang et al., 2011 b)。
大豆可天然固氮和碳,为动物和人类提供营养所需的植物脂肪和蛋白,同时还可兼作饲料和生物能源,是重要的粮油兼用型作物(Thelen and Ohlrogge, 2002; Bhushan et al., 2007)。然而,一些生物胁迫或非生物胁迫致使大豆减产,如病原菌侵染,以及水淹、干旱和盐碱化等(Ohyanagi et al., 2012 )。为选育耐淹、耐旱、耐盐和抗病突变体,蛋白质组学因其可以分离和鉴定出参与某特定生物学过程的各种差异表达蛋白而成为研究热点(Ohyanagi et al., 2012 )。
如Yang等人对花叶病毒侵染的大豆和模拟接种的大豆进行差异蛋白组学分析,结果显示:分离并鉴定的25个大豆花叶病毒诱导表达的蛋白主要涉及蛋白质降解(degradation)、防御信号途径(defense signal pathway)、活性氧爆发(reactive oxygen)、细胞壁增厚(reinforcement),以及能量代谢和调节(metabolism regulation)(Yang et al., 2011a)。Oehrle等利用双向电泳对大豆的根瘤进行蛋白组分析,结果显示,鉴定到的蛋白质按功能分主要涉及四大类:第一类是碳代谢(carbon metabolism)相关蛋白;第二类是氮代谢(nitrogen metabolism)相关蛋白;第三类是活性氧代谢(reactive oxygen metabolism)相关蛋白;第四类是膜泡运输(vesicular trafficking)相关蛋白。尤其是膜泡运输相关蛋白的多个、大量表达预示着大分子物质运输和膜蛋白交换活动在根瘤中非常活跃地进行着(Oehrle et al., 2008)。
Ahsan研究小组对不同发育时期大豆的叶片和花蛋白进行分析和鉴定,得到26种不同的叶器官特性蛋白质;再结合免疫共沉淀结果显示,分子伴侣60、热激蛋白70和ATP合酶在组织中持续表达;花器官特异表达的20多个蛋白点按功能分主要涉及线粒体蛋白的运输和装配(transport and assembly)、次生产物代谢(secondary metabolism)和花粉管发育(pollen-tube growth)相关蛋白(Ahsan et al., 2009)。Ahsan还研究了臭氧胁迫下叶片总蛋白组和叶绿体蛋白组,高含量的活性氧和脂氧化酶测定结果显示:臭氧胁迫诱导的蛋白表达调节发生在伤害症状呈现之前。双向电泳分离、质谱鉴定到的30个差异表达蛋白主要涉及光合作用,包括光系统I、光系统II和碳同化相关蛋白的下调表达。淀粉含量下降和蔗糖含量上升,预示臭氧胁迫引起光合作用途径中三羧酸循环的碳分配的改变(Ahsan et al., 2010)。
有趣的是,Yamaguchi和Komatsu两个研究小组在大豆应答水淹胁迫时对木质素的调节意见相左。Yamaguchi对苗期(营养生长2期)大豆初生根蛋白组研究表明,水淹胁迫(water stress, flooding stress, 或waterlogging stress)下根伸长受抑制的原因主要是一些异黄酮类物质代谢上调后,引起木质素上调表达(up-regulated)以保护根免受氧化损伤,但木质素积累却抑制了根的伸长生长(Yamaguchi et al., 2010);而Komatsu等对萌发 2 d 再水淹处理 2 d 后的大豆根和子叶下胚轴细胞壁蛋白质组进行分析,结果表明:脂氧合酶(lipoxygenases)、萌发素(germin-like protein)和糖蛋白(glycoprotein)的前体(precursors),以及一种超氧化物歧化酶(superoxide dismutase [Cu-Zn])均呈下调表达状态。通过活性氧和茉莉酸甲酯的下调表达导致了大豆幼苗根部的木质素下调表达(downregulation),以使大豆根组织免受水淹的伤害(Komatsu et al., 2010)。
基因虽然决定了生物体主要的遗传性状,但其相应的蛋白质组具有时空表达特异性。利用基于双向电泳凝胶的蛋白质组学分析的高效性和准确性依赖于分离效果好的2-D凝胶图,如果近似分子量或等电点的蛋白质重叠在一个点上,那即使挖取2倍差异表达蛋白点测序结果也可能是多样而不确定的。因此,分离效果好的2-DE凝胶图获取是蛋白质组研究的必要和首要条件。本研究小组经多次试验,已建立了分离效果好的大豆根、茎、叶总蛋白双向电泳方法,为后期的大豆差异蛋白质组学研究奠定了基础。同时初步分析了本试验条件下大豆根、茎、叶蛋白质特点,包括分子量和等电点基本特征。
1结果与分析
1.1大豆根、茎、叶蛋白双向电泳图
双向电泳凝胶3次重复试验,均获得了分离效果好、重复性好的大豆根、茎、叶蛋白质双向电泳图(图1)。由图1可知,大豆根、茎、叶总蛋白质点在2-DE胶图中的分布有明显差异,根蛋白质点(图1A)几乎是均匀分布在整个胶图上,第一向电泳(IEF)结束后,从IPG胶条的酸端(pH 4)到碱端(pH 7)都有蛋白质点,又经第二向电泳(SDS-PAGE)后一些小分子量蛋白点(接近Marker的最小带14.4 kD)跑到了胶的近底部,其形状又大又圆(图1A, 框1),说明其蛋白浓度高。
|
图1 大豆根(A)、茎(B)、叶(C)蛋白质双向电泳图 Figure 1 2-DE pattern of the proteins of soybean roots (A), stems (B) and leaves (C) |
茎蛋白质点(图1B)多分布在胶图的左中部分,IEF结束后,绝大部分蛋白点分布在了IPG胶的酸端(pH 4~6),较少的蛋白点分布在pH 7处;浓度大的小分子量(<14.4 kD)蛋白质点和大分子量(>66.2 kD)的蛋白质点凝胶近底部并不多(图1B, 框2),说明偏弱酸性的蛋白质在茎中分布较多。
叶蛋白质点(图1C)主要集中分布在凝胶图上的较大分子量(25~116 kD间)区间,尤其是分子量在45~66.5 kD之间的蛋白点又多又大,浓度最高;但浓度大的小分子量蛋白点不多。
1.2大豆根、茎、叶蛋白质分子量和等电点分布特点
将凝胶图打印于A4纸,用直尺沿凝胶图的IEF方向将pH 4~7的长度均分画横线,然后标记pH 4、pH 5、pH 6、pH 7;再用直尺沿凝胶图的SDS-PAGE方向、找到蛋白分子量Marker的条带(116 kD, 66.2 kD, 45 kD, 35 kD, 25 kD, 18.4 kD, 14.4 kD),按Marker的条带的位置画横线,然后在横线和竖线的交叉点标记上蛋白质点的分子量和等电点试验值。同时将以上各交点处蛋白质的分子量和等电点赋值到PDQuest软件中的凝胶图上,即可由软件读出所有蛋白质点的分子量和等电点(试验值),并做简单分析可知:本实验条件下所分离到的大豆根、茎、叶蛋白质点都在1 000~1 300个之间,其分子量和等电点分布特征(图2; 图3)。
.png) 图2 大豆根、茎、叶总蛋白质点分子量的分布 Figure 2 Tendency of Mr distribution of protein from root, stem and leaf of soybean identified by 2-DE |
|
图3 大豆根、茎、叶总蛋白质点等电点的分布趋势图 Figure 3 Tendency of pI distribution of protein from root, stem and leaf of soybean identified by 2-DE |
由图2、图3可知,等电点在6~7之间的蛋白质:根蛋白数目>叶蛋白数目/茎蛋白数目;等电点在5~6之间的蛋白质:叶蛋白数目>茎蛋白数目>根蛋白数目;等电点在4~5之间的蛋白质:茎蛋白数目>根蛋白数目>叶蛋白数目。简言之,大豆幼苗根蛋白质偏弱碱性,茎蛋白偏弱酸性,而叶蛋白质大部分等电点在5~6之间。
从实际的2-DE图谱(图1)和PDQuest软件分析给各蛋白点分子量和等电点赋值后统计结果来看(图2; 图3),苗期大豆的根、茎、叶总蛋白的分子量和等电点分布趋势均有差异。小结如下:(1)大豆根总蛋白偏弱碱性、pI在6~7之间的蛋白点数达总蛋白数的1/3以上;MW<18.4 kD的小分子量蛋白,和MW>75 kD的大分子量蛋白在根中的含量均高于茎、叶;(2)大豆茎总蛋白偏弱酸性、pI在4~5之间的蛋白点数达总蛋白数的1/3以上;25<MW<45的中小分子量蛋白在茎中的含量均高于根、叶;(3)大豆叶中,近中性(pI 5-6)蛋白含量过半(占总蛋白数的62%),其蛋白质多集中在分子量25~75 kD间。
2讨论
大豆的根通常具有合成功能;而茎的韧皮部是有机质的输导组织;叶主要负责光合作用,一些重要的光合相关蛋白就在锚定在叶绿体的类囊体膜上,多数是蛋白复合体。由于其功能不一,根、茎、叶蛋白质显示出不一样的分子量和等电点分布趋势是符合理论预期的。
在利用基于凝胶的蛋白质双向电泳方法对植物应答胁迫的蛋白质组进行解析时,不同科学家对相同材料进行相同处理会有不同的结果(Yamaguchi et al., 2010; Komatsu et al., 2010),甚至同一实验中的同一类蛋白质会检测到上调和下调表达,或者具有相同功能和序列号的蛋白质在2-DE胶上的分布位置大相径庭(蛋白质等电点不一致)(董玉梅等, 2013),不能就一概而论地认为是测序错误或者试验误差。因为蛋白质并不像基因一样稳定存在于生物体中,也不与mRNA一一对应。尽管基因序列决定了蛋白质序列,但蛋白质在执行功能时还可能受到mRNA的转录水平调控、翻译水平调控和翻译后修饰调控等(何华勤, 2011)。以蛋白质翻译后修饰为例,真核生物中可能存在20种以上的修饰类型,比较常见的如糖基化、乙酰化、泛素化和磷酸化等(Taylor et al., 2011; Theillet et al., 2012)。其中,起信号转导的开关作用的磷酸化修饰后,蛋白质分子量几乎不改变,但pH值会有改变,即其蛋白质等电点会改变、则在凝胶中的位置会向左(或向右)移动。因此,当检测到功能和序列相同的蛋白质点在凝胶中的分布位置不一样(等电点不同)时,不都是测序错误或实验误差,而正好是蛋白质修饰多样化、功能多样化的表现之一(董玉梅等, 2013)。
由此可知,重复性好的蛋白质双向电泳方法是蛋白质功能和性质确定的重要保障。揭示植物不同组织、器官的蛋白质功能是揭示生命活动规律和蛋白质作用分子机制的直接和有力手段。结合图1、图2和图3的结果和分析可知,大豆根、茎、叶总蛋白质的分子量和等电点分布有较大差异,由此推测大豆根、茎、叶蛋白质在应答各种生物或非生物胁迫时的表现可能会从不同的角度补充解释大豆抗性的分子机制,因此,本方法的建立是利用大豆根、茎、叶蛋白组解析大豆应答各种胁迫的分子机制的前提、是技术基础。而且,此法所分离到的蛋白点比Aghaei等(2009)人分离的蛋白点(300多个)多,故本研究对进一步利用蛋白质组解析大豆应答胁迫的相关分子机制奠定了基础。
3材料与方法
3.1植物材料的培养、取材及预处理
大豆品种郑9525(市售),播种于装有花卉基质的花盆(直径24 cm),每天12 h光照(25℃)/12 h黑暗(20℃)光照培养室生长,大豆长至两对真叶完全展开,备用。
取材,灭菌剪刀剪取大豆叶片,茎秆剪成约1~2 cm长,迅速置于-80℃冰箱保存备用;然后将大豆的根从基质中小心取出,尽量保持完整,于自来水下冲洗至干净,然后用滤纸将水吸干,再置于-80℃冰箱保存备用。
3.2大豆根、茎、叶双向电泳方法
3.2.1大豆根、茎、叶蛋白样品制备及浓度测定
本研究所用的大豆根、茎、叶蛋白样品破碎方法为液氮研磨(机械)法;蛋白沉淀方法为TCA/丙酮(有机溶剂)沉淀法(何华勤, 2011),具体如下。
取预先灭菌、-20℃预冷的研钵置于冰上,称取预先备好的植物组织(叶片1 g; 根, 茎各1.5 g)液氮研磨后,转入50 mL灭菌的离心管中,每管加入20 mL 10% TCA /丙酮(用前现加 DTT 0.1% 质量体积比 0.1g/100 mL);涡旋混匀,放置在试管架上置于-20℃沉淀过夜(14~18 h)。
取出沉淀,于4℃,20 000 rpm,离心20 min,弃上清收集沉淀;-20℃预冷的80%丙酮(现加DTT 0.1%和PMSF 100 μL/10 mL) 20~25 mL/管,涡旋并重悬沉淀(作用是清洗蛋白),置于-20℃沉淀1~2 h;4℃,20 000 rpm,离心20 min,弃上清,收集沉淀,如此清洗蛋白3次,直至蛋白呈白色沉淀,弃上清,循环水真空泵干燥成粉末,转入灭菌的 2 mL离心管中,于-80℃冰箱保存备用。
蛋白质上样前必须测定并调准浓度。从-80℃取出蛋白质干粉,分别称取30 mg/管(1.5 mL),加入450 μL蛋白水化液(15 μL/mg)充分溶解蛋白粉末,Bradford (1976)法测定蛋白质浓度,以牛血清蛋白制作的蛋白标线斜率均在 99% 以上。如表1,按纵列顺序添加①②③种试剂,分别读出空白对照及1、2、3、4、5、6管牛血清蛋白浓度为0、0.1、0.2、0.4、0.6、0.8、1.0,且斜率在95%以上才说明此蛋白标线制作正确、能用于蛋白浓度的测定。
|
表1 蛋白标线制作试剂表 Table 1 Reagents needed by stand curve of protein |
浓度测定时(见表2),按纵列顺序分别向10 mL灭菌离心管中添加G250、水化液和蛋白样品,测定并读数即可。
因核酸蛋白测定仪上显示的浓度是蛋白被稀释10倍后的浓度,因此,蛋白的实际浓度是读数的10倍。记录好每个蛋白样品的浓度后,按照17 cm 胶条的上样量将蛋白浓度调至600 μg/300 μL。
 表2 根、茎、叶蛋白质浓度测定 Table 2 Concentration of the proteins extracted from roots, stems and leaves of soybean |
3.2.2 IPG胶条溶胀及蛋白质一向电泳(IEF)
将定量好的蛋白溶液300 μL (含蛋白600 μg) 于室温,20 000 rpm,离心20 min,取上清转入水化盘中(此步骤的目的是去除一些不容的杂质),转入IPG胶条,于20℃水化(溶胀)过夜。溶胀完全的胶条转入等电聚焦仪(PROTEAN IEF Cell, BIO-RAD),进行聚焦(表3)。
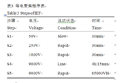 表3 等电聚焦程序表 Table 3 Steps of IEF |
其他参数:GELS : 2;Limit : 50;Temp: 20℃。
3.2.3蛋白质SDS-PAGE
平衡前,按2块胶(17 cm 胶条)的量(150 mL)配制12.5%的SDS-PAGE凝胶液如下:①ddH2O 62.7 mL;②30%丙烯酰胺溶液Acr/Bis 83.3 mL;③1.5 mol/L Tris (pH 8.8) 50 mL;④10% SDS2 mL;Ⅰ10% APS过硫酸氨2 mL;ⅡTEMED 35.6~40 μL。按顺序加入试剂①②③④,用玻棒缓慢搅拌混匀15~30 mim,再加入试剂Ⅰ、试剂Ⅱ,缓慢混匀后即可灌胶,用异丙醇密封胶面。待胶凝固后,倾出异丙醇,改加ddH2O压平胶面,用保鲜膜覆盖,备用。
平衡时,17 cm胶条需平衡液5 mL /根/次。平衡液I现加(DTT, 1%质量体积比),缓慢摇晃平衡15 min;平衡液II现加(IAA, 5%质量体积比),平衡15 min。平衡期间,准备蛋白分子量Marker (使用前沸水煮5 min)。
将胶条和吸有蛋白分子量Marker的滤纸片转入制备好的SDS聚丙烯酰胺凝胶。二向电泳程序,40 mA恒流电泳。看到溴酚蓝指示线整齐泳出后,再将电压调至30 mA/根直到电泳结束。
3.2.4考马斯亮蓝凝胶染色及扫描
电泳结束,需对凝胶中的蛋白质点进行固定,固定液配方(2块胶: 乙醇400 mL, 乙酸100 mL, ddH2O 500 mL, 混匀(1 000 mL),每块胶倒入500 mL固定液,固定3 h;循环水真空泵吸干固定液换上染色液。考染法:2块胶染液配方:ddH2O 200 mL,磷酸100 mL,硫酸铵100 g,G250称取1.2 g,ddH2O定容至800 mL (搅拌30 min),最后加上甲醇200 mL,总体积1 000 mL。染色过夜,直到胶点明晰可辨。二步法凝胶脱色。2块胶脱色液Ⅰ配方:硫酸铵75 g用ddH2O溶解后定容至1 350 mL,再加甲醇150 mL至总体积1 500 mL。其中1 000 mL用于第一次脱色20~30 min;剩余的500 mL脱色液Ⅰ,再加500 mL ddH2O,混匀(1 000 mL),即脱色液Ⅱ。第二次,脱色(液Ⅱ) 12~16 h,或过夜,直至背景颜色透明而蛋白点清晰易辨。于GE Image Scanner Ⅲ扫描仪上扫描,分辨率统一设为300 dpi。
3.2.5凝胶图像的PDQuest 7.4软件分析
PDQuest软件读取2-DE图像,用点细化(Spot segmentation)的方法检测和定量蛋白质斑点,主要包括: 斑点检测和定量、凝胶匹配、相对分子量和等电点的确定、报告输出等。
作者贡献
董玉梅,苏源,余萍共同参与试验全过程,由董玉梅完成论文初稿。梅馨月、杨敏参与部分研究工作。何霞红参与指导工作,李成云负责研究指导及论文修订工作。全体作者都阅读并同意最终的文本。
致谢
本研究由国家973项目“作物多样性对病虫害生态调控和土壤地力的影响”(2011CB100400)资助。
参考文献
Aghaei K., Ehsanpour A.A., Shah A.H., and Komatsu S., 2009, Proteome analysis of soybean hypocotyl and root under salt stress, Amino Acids, 36(1): 91-98
http://dx.doi.org/10.1007/s00726-008-0036-7
PMid:18264660
Ahsan N., and Komatsu S., 2009, Comparative analyses of the proteomes of leaves and flowers at various stages of development reveal organ-specific functional differentiation of proteins in soybean, Proteomics, 9(21): 4889-4907
http://dx.doi.org/10.1002/pmic.200900308
PMid:19862761
Ahsan N., Nanjo Y., Sawada H., Kohno Y., and Komatsu S., 2010, Ozone stress-induced proteomic changes in leaf total soluble and chloroplast proteins of soybean reveal that carbon allocation is involved in adaptation in the early developmental stage, Proteomics, 10(14): 2605-2619
http://dx.doi.org/10.1002/pmic.201000180
PMid:20443193
Bhushan D., Pandey A., Choudhary M.K., Datta A., Chakraborty S., and Chakraborty N., 2007, Comparative proteomics analysis of differentially expressed proteins in chickpea extracellular matrix during dehydration stress, Molecular & Cellular Proteomics, 6(11): 1868-1884
http://dx.doi.org/10.1074/mcp.M700015-MCP200
PMid:17686759
Bradford M.M., 1976, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 72: 248-254
http://dx.doi.org/10.1016/0003-2697(76)90527-3
Colmsee C., Mascher M., Czauderna T., Hartmann A., Schluter U., Zellerhoff N., Schmitz J., Brautigam A., Pick T.R., Alter P., Gahrtz M., Witt S., Fernie A.R., Bornke F., Fahnenstich H., Bucher M., Dresselhaus T., Weber A.P., Schreiber F., Scholz U., and Sonnewald U., 2012, OPTIMAS-DW: A comprehensive transcriptomics, metabolomics, ionomics, proteomics and phenomics data resource for maize, BMC Plant Biology, 12: 245
http://dx.doi.org/10.1186/1471-2229-12-245
PMid:23272737 PMCid:PMC3577462
Dong Y.M., He X.H., Su Y., Li C.Y., Xiao Q.Y., Zhao Z.L., and Zhu Y.Y., 2013, Proteomic Analysis of Glycine max Leaf in Response to Stress of Bipolarismaydis, Fenzi Zhiwu Yuzhong(Molecular Plant Breeding), 11 (5): 476-485 (董玉梅, 何霞红, 苏源, 李成云, 肖秋芸, 赵正龙, 朱有勇, 2013, 大豆应答玉米小斑病菌胁迫的蛋白质双向电泳分析, 分子植物育种,11(5): 476-485)
He H.Q., ed., 2011, Concise proteomics, China Forestry Publishing House, Beijing, China, pp.1-10 (何华勤, 主编, 2011, 简明蛋白质组学, 中国林业出版社, 中国, 北京, pp: 1-10)
Hakeem K.R., Chandna R., Ahmad P., Iqbal M., and Ozturk M., 2012, Relevance of proteomic investigations in plant abiotic stress physiology, OMICS., 16(11): 621-635
http://dx.doi.org/10.1089/omi.2012.0041
PMid:23046473
Komatsu S., Kobayashi Y., Nishizawa K., Nanjo Y., and Furukawa K., 2010, Comparative proteomics analysis of differentially expressed proteins in soybean cell wall during flooding stress, Amino Acids, 39(5): 1435-1449
http://dx.doi.org/10.1007/s00726-010-0608-1
PMid:20458513
Liu Y.Y., 2009, proteomics research of silkworm larva and pupal stage development related protein, Thesis for M.S., Soochow University, Supervisor: Tan J.Z., pp.1-18 (刘艳艳, 2009, 家蚕幼虫期和蛹期发育相关蛋白质组的研究, 硕士学位论文, 苏州大学, 导师:谈建中, pp.1-18)
Oehrle N.W., Sarma A.D., Waters J.K., and Emerich D.W., 2008, Proteomic analysis of soybean nodule cytosol, Phytochemistry, 69(13): 2426-2438
http://dx.doi.org/10.1016/j.phytochem.2008.07.004
PMid:18757068
Ohyanagi H., Sakata K., and Komatsu S., 2012, Soybean Proteome Database 2012: update on the comprehensive data repository for soybean proteomics, Frontiers in Plant Science, 3: 110
http://dx.doi.org/10.3389/fpls.2012.00110
PMid:22661982 PMCid:PMC3362740
Que Y.X., Xu L.P., Lin J.W., Ruan M.H., Zhang M.Q., and Chen R.K., 2011, Differential Protein Expression in Sugarcane during Sugarcane-Sporisorium scitamineum Interaction Revealed by 2-DE and MALDI-TOF-TOF/MS, Comp. Funct. Genomics, doi: 10.1155/2011/989016
http://dx.doi.org/10.1155/2011/989016
Taylor N.L., Heazlewood J.L., and Millar A.H., 2011, The Arabidopsis thaliana 2-D gel mitochondrial proteome: Refining the value of reference maps for assessing protein abundance, contaminants and post-translational modifications, Proteomics, 11(9): 1720-1733
http://dx.doi.org/10.1002/pmic.201000620
PMid:21472856
Theillet F.X., Smet-Nocca C., Liokatis S., Thongwichian R., Kosten J., Yoon M.K., Kriwacki R.W., Landrieu I., Lippens G., and Selenko P., 2012, Cell signaling, post-translational protein modifications and NMR spectroscopy, Journal of biomolecular NMR, 54(3): 217-236
http://dx.doi.org/10.1007/s10858-012-9674-x
PMid:23011410
Thelen J.J., and Ohlrogge J.B., 2002, Metabolic engineering of fatty acid biosynthesis in plants, Metab. Eng., 4(1): 12-21
http://dx.doi.org/10.1006/mben.2001.0204
PMid:11800570
Yamaguchi M., Valliyodan B., Zhang J., Lenoble M.E., Yu O., Rogers E.E., Nguyen H.T., and Sharp R.E., 2010, Regulation of growth response to water stress in the soybean primary root. I. Proteomic analysis reveals region-specific regulation of phenylpropanoid metabolism and control of free iron in the elongation zone, Plant Cell Environment,33(2): 223-243
http://dx.doi.org/10.1111/j.1365-3040.2009.02073.x
PMid:19906149
Yang H., Huang Y., Zhi H., and Yu D., 2011a, Proteomics-based analysis of novel genes involved in response toward soybean mosaic virus infection, Mol. Biol. Rep., 38(1): 511-521
http://dx.doi.org/10.1007/s11033-010-0135-x
PMid:20373035
Yang F., Svensson B., and Finnie C., 2011b, Response of germinating barley seeds to Fusarium graminearum: The first molecular insight into Fusarium seedling blight, Plant Physiol. Biochem., 49(11): 1362-1368
http://dx.doi.org/10.1016/j.plaphy.2011.07.004
PMid:21798752